



PUFAs & The Palisades: Lipid Peroxidation, Oxidative Stress & Cell Death
Health and vitality come from the controlled burn of macronutrients to produce energy. Cellular dysfunction and death result from uncontrolled burns.

Not medical advice.
Large parts of Los Angeles burned to the ground in early 2025. In theory, LA had the material resources and infrastructure necessary to prevent or limit this type of disaster. Despite this, fires of Biblical intensity burned the Palisades and other parts of LA to the ground. The city’s resource management systems were not in healthy balance. Critical protective processes were starved of energy, and burnable material was allowed to pile up. With help from the Santa Ana winds, what should have been a minor nuisance was catalyzed into a hellish nightmare.
What can this apocalyptic catastrophe teach us about biology and human health?

Three things were out of balance in Los Angeles, allowing small and isolated fires to transform into roaring behemoths:
Multiple flame-starters ignited small fires. These would have remained local and been easily contained, but…
Due to poor city management, dry brush and other readily burnable material accumulated near homes and other buildings. In addition…
Fire-fighting capacity was limited: water reservoirs were dry and the fire department was impotent. The LA government had not invested adequate resources (energy) into these protective mechanisms.
The average person today manages their body like LA’s government has managed Los Angeles. In the body, Reactive Oxygen Species (ROS) are your flame-starters. Antioxidant capacity is your fire-fighting potential. Readily burnable materials are the components of your cells that can be easily ignited (oxidized) by ROS.
In this analogy, oxidative stress occurs when cell biological fires burn beyond the capacity to properly manage and contain them. Combustion is a natural consequence of oxygen-based metabolism. ROS production cannot be avoided. When cells are metabolically healthy, small burns can be easily repaired, and fires contained.

Responsible citizens of LA use small, well-managed fires every day to cook their food and do other useful work. Likewise, our cells detect patterns of ROS production to help ensure proper cellular upkeep. Collateral damage is minimal when systems are in check—not too much ROS production (flame-starters), not too little fire-fighting capacity (antioxidant capacity), and no excess dry brush (readily oxidizable material) lying around. Storing some dry wood in the shed is fine, but you don’t let it pile up in every room of the house or the alleyway between you and your neighbor’s home.
Many factors determine the balance of all these things—our light environment, food environment, and pattern of bodily movement through time and space—shaping our health and disease potential. Because we’re eukaryotic organisms, most of our energy is produced from social networks of mitochondria within our cells. As a result, when it comes to ROS production, lipid peroxidation, and activation of cell death pathways, a lot of the action takes place within and near these organelles.
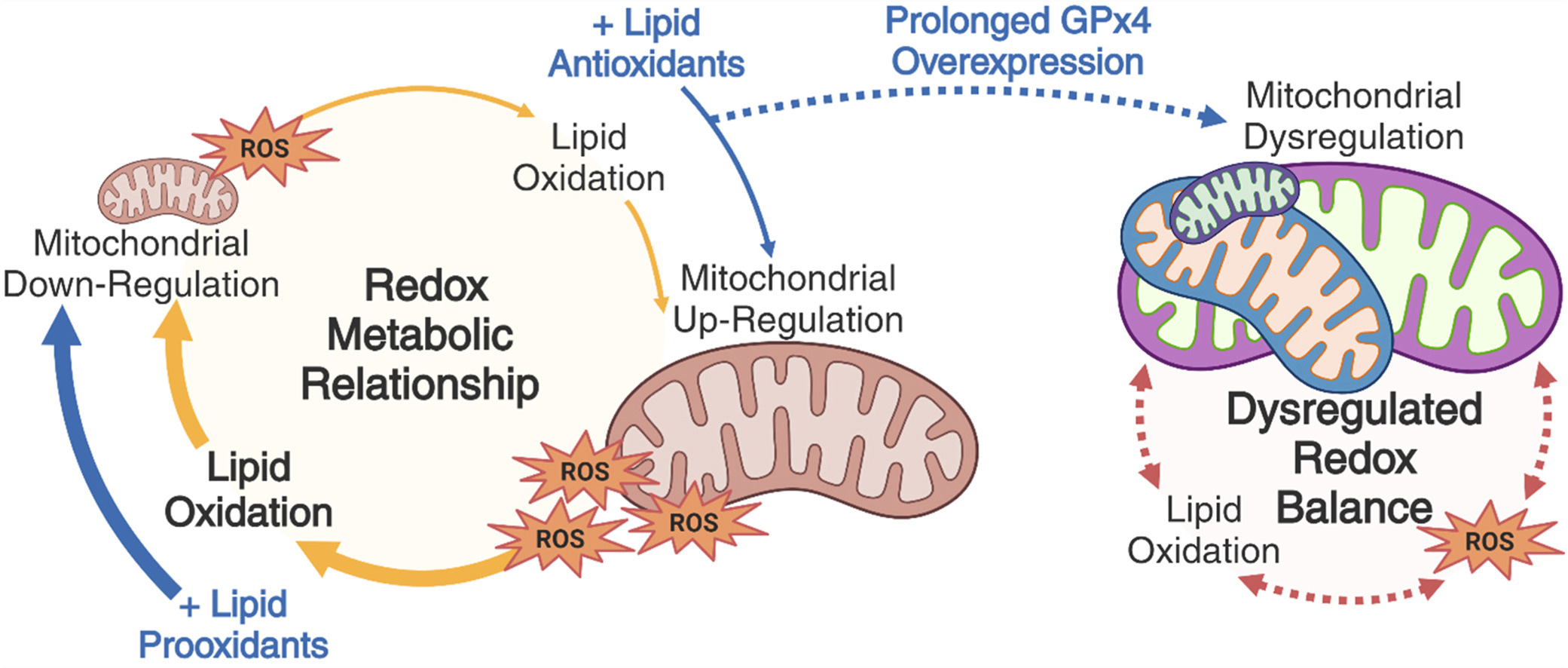
The LA fire analogy may not be perfect, but it should help those new to thinking about oxidative stress comprehend some key pieces of cell biology. In this post, we will focus largely on specific components of our diet, polyunsaturated fatty acids (PUFAs). These fatty acids play the role of readily burnable material and influence the potential for a small kitchen fire to set the house—and even the whole neighborhood—on fire.
What is lipid peroxidation?
If burnable material is anything that can be oxidized, then PUFAs are dry kindling, perfectly suited to starting a fire. Lipid peroxidation is the oxygen-dependent degradation of PUFAs. ROS can ignite them. When they burn, toxic byproducts are released. If the fire cannot be contained or put out, this can result in cell death.
Lipid peroxidation is akin to fire itself. The fire’s heat and extent of spread are constrained by the density of PUFAs in lipid membranes—how much dry kindling has been allowed to build up and whether it has piled up near key cellular components. The balance of ROS production and antioxidant capacity depends on multiple factors. The more the balance is tipped in favor of ROS production, the more chances there are to ignite a piece of dry wood (PUFA). The bigger the fire, the more heat and toxic fumes.

There are three basic phases of the lipid peroxidation cascade:
Initiation: An ignitor needs to start a fire. ROS plays this role.
Propagation: Once a fire starts, it tends to keep burning as long as fuel is present. Lipid peroxidation produces reactive products that can set off chain reactions.
Termination: Fires end when they either run out of fuel or are actively put out by firefighters (antioxidants).
For one reason or another, small fires sometimes turn into big fires, and the whole house burns down (cell death). Some houses are well-insured and can be rebuilt. Other cell types, such as most neurons, cannot—destruction yields no insurance payout, and property values in the whole neighborhood decline (neurodegenerative disease).
I used this basic analogy in my conversation with Dr. Pamela Maher, discussing her research on how reactive oxygen species (ROS) affect lipid peroxidation and a form of cell death known as ferroptosis.
If you want to brush up on the basics of dietary PUFAs and related biology or start learning about how things like light influence oxidative stress, try these resources before reading on:
To learn about lipid biology, dietary PUFAs, and related topics, try these resources:
Article: Eating Fat Like Never Before
Article: Biological Consequences of Specific Dietary Fatty Acids
To learn about photobiology and the effects of light on health and oxidative stress, try these resources:
Podcast: Photobiology, Sunlight, Firelight, Incandescent Bulbs vs. LEDs, Mitochondria, Melatonin, Sunscreen & the Optics of the Body | Scott Zimmerman
Podcast: Benefits & Risks of UV Radiation & Sunlight, Skin Health, Vitamin D, Nitric Oxide, Evolution of Skin Color | Richard Weller
Lipid peroxidation & cell death
Cell death is a common part of cellular life. Some cell types divide and die off frequently. Skin cells, for example, have a high turnover rate. Some never divide once they reach their state of terminal differentiation (final form). Neurons are like this. With special exceptions, when a neuron dies, it’s over. When someone loses enough of their dopamine neurons, we call it Parkinson’s Disease, and those cells are not coming back.
In other cell types and contexts, cell death is good. Cell death is a welcome outcome if an easily replaceable cell becomes cancerous or infected. Cancer cells pop up often, and pathogens frequently cross our cellular borders. In most cases, these things do not become a problem for the whole organism, and we don’t even notice—problematic cells are detected, killed, and replaced.
Cell death is also a natural part of development. Without properly regulated cell death, we would have webbing between our fingers and other malformations. When building and developing a city, you often want to bulldoze infrastructure that’s no longer needed, making room for healthy new developments. But you don’t want fires to spring up and burn uncontrollably.
Learn more about oxidative stress:
Podcast: Cell Death, Oxidative Stress, PUFAs & Antioxidants | Pamela Maher
Podcast: Cellular Aging, Oxidative Stress, Antioxidants, Life Extension & Health Supplements | Leonard Guarente
The bottom line is that cell death is not inherently good or bad. It is a regulated process. In certain cellular contexts, it’s normal and healthy. In other contexts, it can be triggered in irreplaceable cells, with loss resulting in health deficits. If cells become dysregulated in ways that impair their ability to detect and handle stress, cell death mechanisms can be triggered even when it’s not really beneficial.
We are not going to get into much of the hardcore details of cell death and oxidative stress. Just know these core things, and consult the resources below if you want to learn more:
There are multiple forms of cell death, differing in their molecular and cellular details.
Reactive oxygen species (ROS) are an inevitable consequence of oxygen-based metabolism. Mitochondria produce and sense ROS as a normal part of their critical role in energy production.
In a healthy cell, ROS production and antioxidant capacity are in a balance that prevents normal levels of ROS production (“small flames”) from triggering unwanted cell death (“burning down the house”).
Polyunsaturated fatty acids (PUFAs) are readily oxidizable (“burnable") by ROS because they contain multiple double-bounds. PUFAs always exist in the lipid membranes of our cells, but their density can change based on dietary fat intake.
Lipid peroxidation happens when lipids such as PUFAs are oxidized, producing toxic byproducts like malondialdehyde (MDA) or 4-hydroxynonenal (4-HNE), which can damage the cell or trigger cell death. These compounds are the chemical stressors underlying oxidative stress.
The extent of lipid peroxidation depends on the density of PUFAs within a cell. A “fire” may be small and put out before it causes irreparable damage. But if there’s dry kindling everywhere, it may burn down the house or even spread to a neighbor.
Lipid peroxidation is a core component of most or all known forms of cell death.
Increased ROS production is often insufficient to trigger unwanted cell death—cell death can be experimentally blocked by preventing the lipid peroxidation downstream of ROS production. In other words, excess lipid peroxidation is often the final cause of cell death.
Here is a nice graphic from this review paper, which gives a sense of the different forms of cell death and the pathways that trigger them. Despite differences in their molecular details, the final outcome (cell death) is the same, with lipid peroxidation playing a key role in the chain of events.

Reactive oxygen species (ROS) can be formed in many ways. A major source is always mitochondrial respiration because oxygen plays a key role in energy production for creatures like us. Other aspects of our internal metabolism can also produce ROS, as does exposure to external forces like UV radiation and chemical oxidants.

Similar to a macroscopic fire, the extent of the fire ultimately depends on key factors that regulate its initiation, propagation, and termination. Again, think in terms of ROS production, PUFA density, and antioxidant capacity—flame-starters, burnable material, and fire-fighting capacity.

Recall that our cell membranes are phospholipid bilayers. Every phospholipid in the membrane contains a hydrophilic “head” and a fatty “tail” consisting of two fatty acids. Each fatty acid can be saturated, monounsaturated, or polyunsaturated, depending on whether it contains zero, one, or 2+ carbon-carbon double bonds. Each one can be either type. Only unsaturated fatty acids (MUFAs or PUFAs) can be oxidized, and the more double bonds fatty acids have, especially at certain locations (see below), the more readily they can be oxidized.
As you might imagine, the more unsaturated fatty acids (especially PUFAs) in the plasma membrane, the more places there are for ROS to start a fire. As we’ll see below, the fat composition of your diet can change the fatty acid content of your cells, and the PUFA content of people’s fat tissues has been rising for decades. This is akin to the dry brush that wasn’t being tended to in Los Angeles—it built up along roads and between buildings, enabling fire to easily jump from house to house.


Recent work has shown that this analogy is more accurate than you might think: the higher the PUFA density in membrane phospholipids, the more prone they are to undergo ferroptosis, a form of cell death. Lipid peroxidation (“fire”) can even jump from one cell to another. One minute, your house catches on fire; next, the whole Palisades neighborhood burns.

So, having a high PUFA density in the cell membrane can make a cell more prone to ferroptosis, which can then spread to adjacent cells through membrane contacts. The more PUFAs in those cell membranes, the more “flammable” they are. If increasing PUFA density increases your risk of a lipid peroxidation “fire,” does reducing it have the opposite effect?
As it turns out, cells can be made more “fire resistant” by swapping out PUFAs for other, less readily oxidizable fatty acids. Exposing cells to exogenous monounsaturated fatty acids (MUFAs) displaces PUFAs from membranes, promoting a ferroptosis-resistant cell state. In other words, the balance of fatty acids determines how readily a cell will undergo ferroptosis.
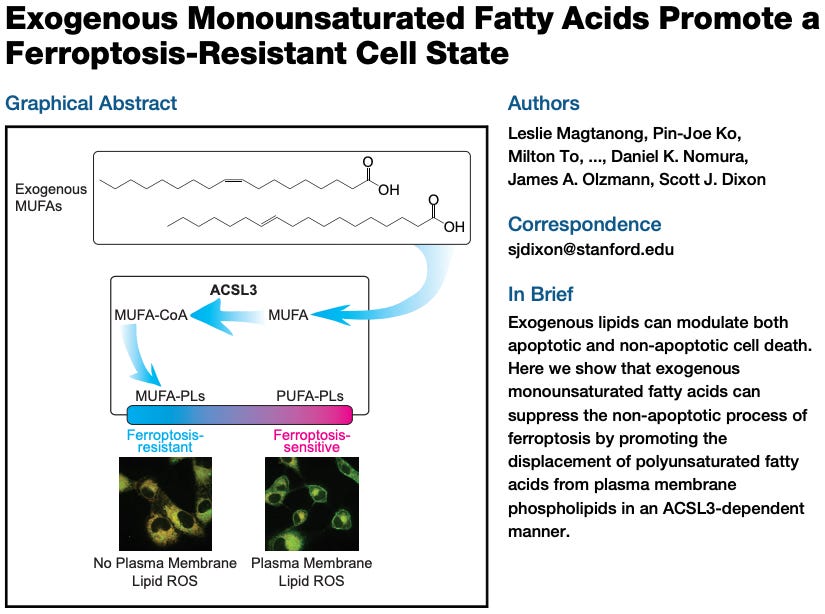
Studies like the above are done in vitro (cell cultures in a dish), enabling scientists to exercise far more precision and control than can be done in live animals. Technical limitations often prevent things at this scale from being measurable in vivo, raising the question of the extent to which these phenomena happen inside us. As we will see, there is no good reason to doubt that these things occur in vivo. We know that ROS production is an inevitable consequence of mitochondrial metabolism and lipid peroxidation can and does happen, producing cytotoxic byproducts.
We also know that dietary PUFA intake, especially of ω-6 PUFAs found in seed oils and processed foods, has risen for over a century. Over that period, people have not only packed on more fat, but the concentration of PUFAs in their fat has gone up.
To learn more about PUFAs, oxidative stress, and cell death, check out these M&M episodes:
Podcast: Cell Death, Oxidative Stress, PUFAs & Antioxidants | Pamela Maher
Podcast: Seed Oils, Omega-6 PUFAs, Inflammation, Obesity, Diabetes, Chronic Disease & Metabolic Dysfunction | Chris Knobbe
How Has the PUFA Content of Fat Tissue Changed Over Time?
In this article, I unpacked how human fat consumption has changed over time. One of the major patterns in history is that PUFA intake, especially ω-6 PUFAs, has increased since industrialization. Linoleic acid (prominent in seed oils) is the single most abundant ω-6 PUFA in the modern food environment. Prior to the 20th century, linoleic acid intake rarely represented more than ~1-2% of calorie intake. By 2008, linoleic acid alone accounted for almost 12% of the calories in standard Western diets. As far as I’m aware, ω-6 PUFA intake is the only macronutrient that has more or less monotonically increased, on average, since industrialization began. Intake of other macronutrients—saturated fat, trans fat, sugar—has waxed and waned in different ways, but ω-6 intake has pretty much moved in one direction.

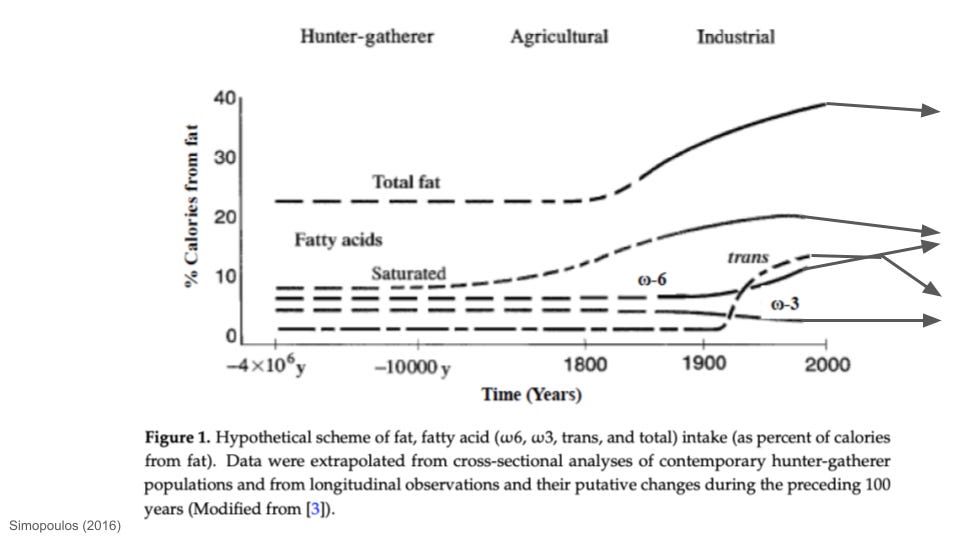
Obesity rates have obviously exploded in parallel to the rise in ω-6 intake. People have not only gotten fatter, but the density of linoleic acid in fat tissues has risen dramatically. Biopsies from fat tissue reveal that the linoleic acid of human body fat doubled between the 1950s and early 2000s, in close association with linoleic acid intake.
(Side note: this post is focused on lipid peroxidation and inflammation, but in addition to serving as precursors to pro-inflammatory lipids, ω-6 PUFAs are also precursors to endogenous cannabinoids. What happens when endocannabinoid levels go up? Metabolic changes that promote feeding and obesity.)


As far as I’m aware, this is by far the highest PUFA content observed in the adipose tissue of any animal in recorded history. Even if we knew nothing else about PUFA biology, such as the connections to oxidative stress reviewed above, this is an evolutionarily unprecedented situation. For almost all of human history, people did not have as much body fat as they do today, and the fat they did have has nowhere near the PUFA density seen in people today.
Lipid peroxidation is obviously a problem for cells. Two of the more common and well-studied toxic byproducts of PUFA oxidation are 4-HNE and MDA (mentioned above). Guess where else you find toxic aldehydes and similar compounds like these? Cigarette smoke. Think about it: those french fries you ate were boiled in PUFAs.
But even the normal metabolism of non-deep fried ω-6 PUFAs is an issue—they serve as precursors to lipid mediators of inflammation, such as prostaglandins (aspirin and other NSAIDs inhibit prostaglandin production). As described more in this article and this podcast, modern ω-6 PUFA consumption is high in both absolute terms and relative to ω-3 PUFAs. For most of human history, ω-6 and ω-3 PUFA intake was approximately equal. This is the ideal balance, as ω-3 and ω-6 tend to have opposed effects on inflammation (and other things; see below).


It should be no surprise that fat tissue itself is inflammatory. People are filled with more fat than ever, and it contains the highest density of ω-6 PUFAs in known history—fatty acids that serve as precursors to inflammatory lipids and which make cells prone to lipid peroxidation “fires.” In my view, it’s also no coincidence that more and more new research is showing that certain cancers, such as colon cancer, have their roots in inflammatory states of lipid dysregulation that can be directly linked to dietary ω-6 PUFAs and seed oils.
Learn more about the latest science linking dietary ω-6 PUFAs to cancer:
Podcast: Dietary Fats & Seed Oils in Inflammation, Colon Cancer & Chronic Disease | Tim Yeatman & Ganesh Halade
Paper: Integration of lipidomics with targeted, single cell, and spatial transcriptomics defines an unresolved pro-inflammatory state in colon cancer
Paper: Direct sensing of dietary ω-6 linoleic acid through FABP5-mTORC1 signaling
If a high level of ω-6 PUFA intake and unbalanced ω-6/ω-3 is pro-inflammatory, then we would expect that boosting people’s ω-6 PUFA intake should drive more inflammation. So if we perform a randomized controlled control (RCT), randomly assigning one group of patients to consume more ω-6 PUFAs than the other, we should see inflammatory markers go up, right?
As many health influencers and pro-PUFA members of the heart mafia have pointed out, there aren’t many human clinical trials out there showing that ω-6 PUFA increases inflammation.
What’s going on?
Why Aren’t There More Clinical Trials Showing PUFA Consumption Drives Inflammation?
The short answer is that the randomized controlled trials (RCTs) people can point to usually have not been conducted in a compelling way. Just because someone conducts a human RCT does not mean it was appropriately designed or executed. The clinical trials that have been done often suffer from one or more critical issues, such as:
The trials don’t last long enough. The lipid-driven changes that drive chronic inflammation and obesity (see below) take time to unfold. No one is funding or executing RCTs that last for years or decades, with careful control of diet. That said, the Minnesota Coronary Experiment (1968-73) found that replacing saturated fats with PUFAs (linoleic acid) offered no mortality benefit, despite lower serum cholesterol.
They typically only measure serum markers of short-term injury- or infection-induced inflammation rather than chronic inflammation. The PUFA-based biology we looked at above takes place largely in adipose tissue, where there’s the highest fatty acid density. However, most RCTs only measure a limited number of serum markers from the blood.
The controls and baseline measurements needed to properly analyze and sort data are not done. For example, if you don’t know the baseline ω-6:ω-3 fatty acid tissue composition and intake patterns people have, the ability to detect relevant changes is hindered, assuming the right markers in the right places are measured in the first place (see above).
When people argue about the health effects of ω-6 PUFAs from seed oils, they often fall into one of two camps: those that point to the basic biology (e.g. the things we reviewed above), concluding that high ω-6 PUFA loads will drive inflammation and other negative outcomes. On the other side are people who point to a lack of human RCTs showing this.
I posed this issue to two lipid biologists on M&M 200, in a discussion about their recent study linking PUFA-driven lipid dysregulation to colon cancer:
NJ: There’s basic research, clinical research—different types of studies that different types of scientists conduct—they all have their strengths and weaknesses.
Some people say, “Well, the seed oils are clearly pro-inflammatory because we know all of this biochemistry that's been worked out… but then other people will say… “That's all great. The basic biology sounds good in theory, but we don't see our randomized control trials clearly showing that seed oil consumption causes an increase in all of our favorite inflammatory markers.”
How do you guys think about those elements of the literature?
Dr. Tim Yeatman: Some of these trials I've read [might say], “We did 10 weeks of an obesity diet for patients, and 10 weeks [with] a normal diet. And then we looked at plasma levels of omega-6 and omega-3.” I 10 weeks a lifetime? No. I mean this is glacial stuff. It doesn't happen overnight. It takes decades. Decades of exposure may cause these risks. So, I think some of these studies that are [RCTs]—you can't do it long enough to see the effects—that's number one.
Number two… why do we see an increase in colon cancer under the age of 40? There's a substantial increase in the last 10 to 20 years. So what's what has changed? The only thing that has changed is really, quite frankly, diet and maybe levels of exercise and so forth. But children today have been exposed to more of these PUFAs, these polyunsaturated omega-6 varieties, than I was as a child growing up. So there's a big difference, and we haven't explained it yet clinically— no one has, and we need to figure it out…
… Studies can be manipulated in lots of different ways. That’s why I like the hard science of lipidomics and tissue spatial transcriptomics and the things we did, as opposed to [just] dietary histories [a subjective measure commonly used in epidemiology and human clinical work]. I mean, how really carefully can you take a dietary history on someone?
Thinking about human RCTs or preclinical studies in isolation is simply not wise. In my view, one must take an integrated view and assess the extent to which these types of studies, from basic animal research to human RCTs, fit together (or not). On the issue of dietary PUFAs in human health, it is clear (to me, at least) that the apparent lack of RCTs showing certain outcomes is simply because the appropriate RCTs have not been conducted (and likely won’t be due to practical limitations).
Consider Table 2 from this review paper on PUFAs, which lays out important critical factors that are typically not accounted for in clinical studies:
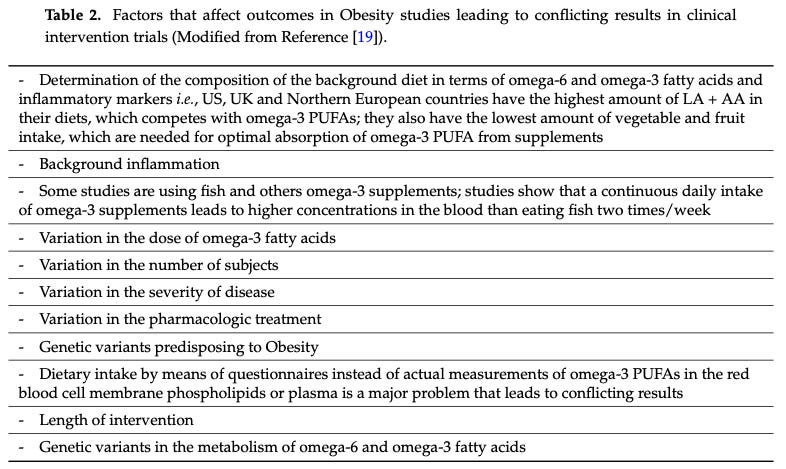
People's background PUFA intake, fat tissue composition, and fatty acid metabolism vary greatly. The genes encoding fatty acid desaturases, which metabolize PUFAs, are also highly variable. There is evidence that, following the transition to agriculture, there was strong natural selection on fatty acid metabolism. Sedentary farming drastically changed the food environment people lived in, including their ω-6 PUFA intake. The specific genetic variants you have influence things like your LDL cholesterol levels, likely playing a role in the extent to which ω-6 PUFAs affect your biology. However, clinical studies pretty much never account for these critical differences between individuals.
Learn more about the evolution of human diet & metabolism:
Podcast: Evolution & Genetics of Human Diet, Metabolism, Disease Risk, Skin Color and Origins of Modern Europeans | Eske Willerslev
Article: What Did Humans Evolve to Eat?
Anyways, let’s say you’re convinced that dietary PUFAs drive tissue inflammation and set you up for lipid peroxidation-driven cellular dysfunction. If you’re carrying more fat tissue than you’d like, and your PUFA load is high, is there anything you can do to help protect against lipid peroxidation and prevent cell damage? Below, I will describe some of the work of Dr. Pamela Maher on this front, which we discussed on M&M 220.
Potential Ways To Prevent Lipid Peroxidation-Induced Cell Death
If you buy the basic story stitched together in this article, then you will want to reduce PUFA intake, the total amount of excess fat tissue you’re carrying, and the risk of cellular damage from peroxidation of the membrane PUFAs already inside you. This will involve doing things to ensure you’re ROS production is in balance with your antioxidant capacity (which we will not be covered here).
There are natural compounds that appear to inhibit or reduce the extent of lipid peroxidation and protect against neurodegeneration. Dr. Maher is studying a variety of these in her lab, which includes compounds derived from strawberries, turmeric, and cannabis. For example. cannabinol (CBN) is a non-intoxicating cannabinoid found in some marijuana products. I won’t go into great detail here, but Dr. Maher’s work shows that CBN can inhibit cell death (ferroptosis) through direct effects on mitochondria. This research is preclinical but suggests that compounds like CBN may have protective effects, helping to maintain good mitochondrial function and activating endogenous antioxidant systems.

The more adipose tissue you have the higher your overall PUFA load, the pressing the need to protect your cells from the toxic effects of lipid peroxidation. Based on the tissue data we saw above, as well as knowing the basic dietary PUFA intake rates of modern people today, there are a lot of obese people walking around with high membrane PUFA levels, placing them at high risk for lipid-derived inflammation and cell dysfunction. Given that GLP-1-based weight loss drugs like Ozempic are now helping many people reduce body in relatively short order, one wonders if those with a high PUFA load might experience high levels of lipid peroxidation-induced inflammation and cell death as a consequence of quickly burning that fat.
Learn more about GLP-1-based weight loss drugs and how they work:
Podcast: GLP-1, Weight Loss Drugs, Ozempic, Obesity, NMDA Receptors, Metabolism & Brain Health | Christoffer Clemmensen
Podcast: Adipose Tissue & Body Fat: Obesity, Insulin, Leptin, Fertility, Weight Loss & GLP-1 Drugs | Sean Hartig
Integrative Thinking & the “Seed Oil Debate”
For me, the "seed oil debate" is a litmus test for someone's ability to think in a critical, integrated fashion across the spectrum of preclinical to clinical research. Understanding basic biology is key not just from a mechanistic point of view but also because it reveals why certain clinical results in humans are missing.
In my experience, some people develop a kind of simplistic “clinical chauvinism” that sometimes allows them to dismiss preclinical research out of hand. People with this mindset are prone to mistake the absence of (clinical) evidence for evidence of the absence. Of course, bias can exist in the opposite direction as well. There are many examples of preclinical results that fail to hold up in human studies. All forms of study have their strengths and weaknesses.
Simply looking at published papers and tallying up the reported results is not enough, especially with the clinical literature. If clinical studies showing an effect are absent from the literature, this could mean that there is no effect there to be observed. It can also mean that proper trials have not been done or that existing RCTs have not been designed to capture the relevant biology—for example, clinical studies measuring one inflammatory marker in serum instead of the relevant lipid-based inflammatory mediators of inflammation from tissue samples—harder to do, so it’s less likely to actually get done.
To give just one example, studies like this are based on analysis of subjective and unreliable data. Even if the data were quantitatively reliable, they look at just a single serum marker of acute inflammation—no direct measurements of adipose or other tissues and no way to control for key variables listed above, such as baseline tissue PUFA density, genetic variation in PUFA metabolism, and so forth.
From feeding and weight gain to cancer, the preclinical research indicates that lots of important biology is happening slowly, on timescales longer than what will get picked up in most clinical trials. The health consequences of diet patterns may take years to manifest in easily measurable clinical endpoints in people. In order to detect effects that are observed over the course of weeks in rodents (see below), you'd need to run a very long clinical study in humans (years). This just isn't going to get done. Should we count that as a "lack of clinical evidence," or recognize that practical limitations prevent clinical trials from detecting "slow" effects like this?
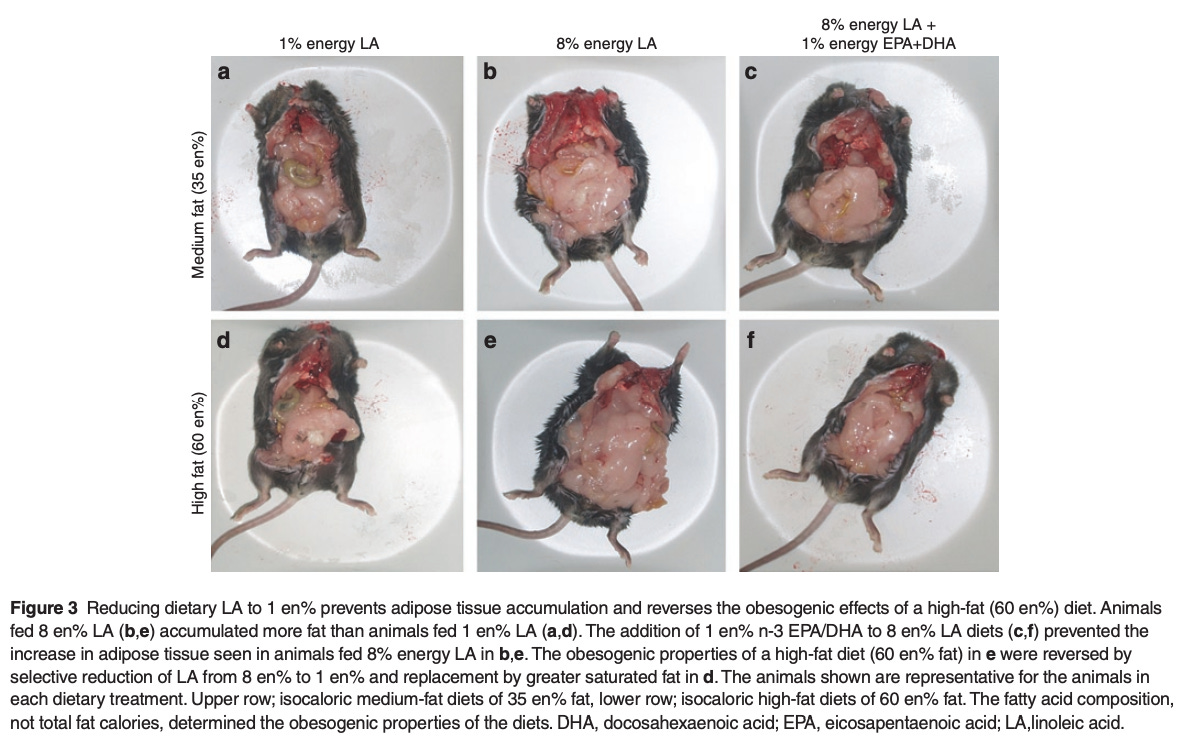
Cancer is another good example. New preclinical research linking ω-6 PUFAs to multiple cancer types has been coming out. But the nature of the underlying biology means that we can't do a "gold-standard RCT" to test this directly in humans. If PUFAs like linoleic acid can promote oncogenesis via chronic tissue inflammation, a tumor obviously isn't going to spring up overnight. These things likely take years to manifest in humans. No one is going to fund or execute a multi-year human clinical trial carefully controlling diet and measuring tissue PUFA composition.
Some of the latest research linking ω-6 PUFAs to cancer:
Paper: Integration of lipidomics with targeted, single cell, and spatial transcriptomics defines an unresolved pro-inflammatory state in colon cancer
Paper: Direct sensing of dietary ω-6 linoleic acid through FABP5-mTORC1 signaling
The human body has never in the natural history of our species been exposed to the PUFA levels we commonly see in people today. For millions of years, our human and proto-human ancestors survived in almost every natural food environment on Earth. One dietary pattern all of those people shared in common: a much more balanced ω-6:ω-3 PUFA intake than present-day humans, with PUFAs never representing more than roughly 2% or so of calories.
It was only ~12,000 years ago, with the dawn of large-scale sedentary farming, that some ancient humans first began consuming modestly higher levels of ω-6 PUFAs. It was only since industrialization, roughly the last century, that ω-6 PUFA intake rose dramatically to become a dominant global dietary pattern.
Ask yourself: what are the odds that our bodies will be metabolically well-adapted to handle a dietary pattern never before seen in natural history? And isn’t it strange that these things (seed oils) only became labeled as “heart healthy” after private corporations amassed a huge surplus of industrial seed oils, and then started paying “health organizations” like AHA to designate them as such?
Los Angeles was poorly managed for years before the right set of conditions sparked one of the worst fires in modern history. You might not have any obvious issues today, but if you let PUFAs accumulate to unnaturally high levels in your body, your cells could end up like the Pacific Palisades…